New paddling submarine needs no engine or power supply
08/17/2018 / By Zoey Sky
According to a report, a group of scientists has created a unique paddling submarine, which doesn’t need an engine, a power supply, or propellant, using only 3D.
The researchers from ETH Zurich, who were led by professor Kristina Shea, collaborated with researchers from the California Institute of Technology (Caltech) in Pasadena, California, to develop a new propulsion concept for swimming robots. The robot “swims” using water temperature fluctuations for propulsion.
The researchers reported the paddling submarine via the journal PNAS.
How the paddling submarine was made
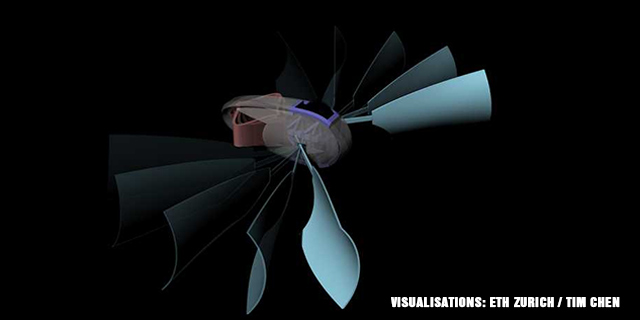
The collaborative team designed a mini-submarine with paddles as a proof-of-concept study. The device measures only 7.5 cm, and it was made with only a multi-material 3D printer.
The paddles move with the help of a bistable propulsion element that is activated by two shape memory polymer strips, which Professor Shea earlier developed with her doctoral student Tim Chen. The polymer strips were designed to expand in warm water, and they help power the robot by acting as its “muscles.”
Once the mini-submarine floats on heated water, the “muscles” expands, making the bistable element snap quickly. This then triggers a paddle stroke. The directional motion, force, and timing of the paddle strokes are “precisely defined by the robot’s geometry and material.”
Each actuating element can only perform one paddle stroke, after which it has to be manually reprogrammed. But the researchers added that new complex swimming robots could be made using multiple actuators.
The mini-submarine developed by the researchers can paddle forward with a single stroke. The swimming robot can also release its “cargo,” which is represented by a coin, then it returns to the starting point with a second paddle stroke in the opposite direction. All of this is done as the robot “senses” changes in the water temperature.
The scientists shared that changing the geometry of the polymer muscles will let them determine the sequence that triggers the paddle stroke. Another thing to note is the thin polymer strips heat up faster in warm water, meaning it responds more quickly than thicker strips. (Related: Researchers create robot fish that can swim right next to real ones in coral reefs.)
The researchers posited that a potential development for the swimming robot includes using polymers that, instead of reacting to the water temperature, respond to other environmental factors like the acidity or salinity of the water.
Shea explained that thanks to the collaboration, the researchers were able to develop “a new and promising means of propulsion that is fully 3D printed, tuneable and works without an external power source.” She concluded that the swimming robot could also be developed to produce a low power device that can be used for ocean exploration.
Fast facts on the temperature of ocean water
The swimming robot moves when the polymer strips react to water temperature. But how does the temperature of water in the ocean vary?
- Earth is heated by solar radiation or incoming energy from the sun. Since the planet is round, the angle of the surface relative to incoming solar radiation often changes depending on the latitude.
- In the tropics, or at low latitudes, direct overhead sunlight received throughout the year warms surface waters.
- Meanwhile, at high latitudes, ocean waters receive less sunlight. For example, both the North and South poles only get around 40 percent of the heat that the equator does. Since the surface waters in the Arctic and Antarctic aren’t warmed as much, they are extremely cold. Sometimes, the surface water even turns to ice.
- Since solar energy is quickly absorbed or reflected in the ocean’s upper surface, only a fraction of solar radiation reaches deeper water, most of which remains cold. Another factor is the fact that water is less dense than cold water. This means cold water sinks while warm water floats above it.
You can read more articles about robots and AI like this swimming robot at Robots.news.
Sources include:
Tagged Under: 3D printing, computing, inventions, machine, mini-submarine, ocean exploration, oceanography, paddling submarine, propulsion concept, robots, shape memory polymer strips, swimming robot, tech, temperature fluctuations, weird science
RECENT NEWS & ARTICLES